Agreement between a sequence-only prediction and the intended
design provides evidence that the sequence encodes the proposed
fold. It does not establish soluble expression, experimental
stability, binding affinity, specificity, or biological function.
Structural disagreement is still useful. It suggests that the
proposed sequence does not robustly encode the intended state, or
that the models disagree about which state is most plausible.
Campaign Foundations · Foundation 3
Computational Design Stages & Tradeoffs
How computational protein-design campaigns move from a prepared target to a diverse, interpretable, and experimentally actionable candidate portfolio.
Computational design is a sequence of decisions
The previous foundation page placed computational design within the larger campaign funnel. This page looks inside that design phase. The goal is not simply to pass structures through a chain of models. It is to use distinct sources of evidence to generate, challenge, compare, and prioritize molecular hypotheses.
The exact tools depend on the target, modality, available infrastructure, and maturity of the field. A mini-protein campaign may generate new backbones and then design sequences onto them. An antibody or VHH campaign may preserve an existing framework while redesigning loops or selected residues. An enzyme campaign may begin from a known scaffold and focus on active-site geometry, substrate positioning, or transition-state stabilization.
Despite these differences, the underlying stages are broadly transferable: prepare the molecular system, generate or modify candidates, design compatible sequences, test structural recovery, evaluate the intended interaction, apply more expensive physical or ensemble-based analyses selectively, seek independent evidence, and construct a diverse shortlist.
The models are not interchangeable.
Each model sees a different representation of the problem and carries
different assumptions and failure modes. A model handoff is therefore
not just a file-format transition. It is a change in what information
is available, what is being optimized, and what kinds of errors may
pass silently into the next stage.
Worked implementation: the Volume 1 mini-binder workflow
The diagram below shows how these general stages are implemented in Volume 1. RFdiffusion generates target-conditioned mini-protein backbones, ProteinMPNN proposes compatible sequences, and ESMFold tests whether those sequences recover the intended fold. A second, sequence-only branch uses Boltz-2 to reconstruct the binder–target complex without receiving the original designed pose. Interface analysis, molecular dynamics, and consensus scoring then narrow the candidate set.
This is one implementation of the broader campaign architecture, not a universal protein-design pipeline. Other modalities may substitute different generation, sequence-design, structure-prediction, docking, energetic, or simulation methods while preserving the same decision logic.
Figure 1. The computational workflow used in Volume 1. The upper path tests whether designed sequences recover the intended mini-protein fold. The lower path asks whether a sequence-only complex-prediction model recovers the intended binder–target geometry. Evidence from both paths contributes to interface triage and candidate prioritization.
Stages in a computational design campaign
1. Molecular-system preparation
Every campaign requires a defined molecular starting point. For a structure-based binder campaign, this often means selecting an appropriate experimental or predicted target structure, identifying the relevant biological assembly and conformational state, extracting the required chains, and assessing unresolved regions, alternate conformations, cofactors, ligands, glycans, and other components that may affect the intended interaction.
Other modalities require analogous preparation decisions. An enzyme campaign may need a substrate- or transition-state model. An antibody campaign may require a framework structure and an epitope definition. A multispecific design may require several target geometries and an explicit model of spatial constraints.
Tradeoff — preparation depth
Over-investing in system preparation can delay the campaign before any candidates are generated. Under-investing can embed structural artifacts or incorrect biological assumptions that propagate through every downstream stage. The objective is not a perfect representation of molecular reality, but a defensible starting model with assumptions that are documented and revisited when necessary.
2. Candidate generation
The generation stage proposes new molecular hypotheses. Depending on the modality, this may involve creating entirely new backbones, sampling loop conformations, docking known scaffolds, mutating an existing sequence, redesigning an active site, or combining known functional elements into a new architecture.
In Volume 1, RFdiffusion generates novel mini-protein backbones positioned against the target surface. The model is conditioned on selected target residues, binder length, and geometric constraints. At this stage, the outputs are candidate backbone geometries rather than complete protein sequences.
Tradeoff — exploration versus exploitation
Imagine a fishing boat on open water. Fish are concentrated in only a few regions, but those regions are not known in advance. Do you fish one location intensively, or distribute your hooks across several locations to map the landscape?
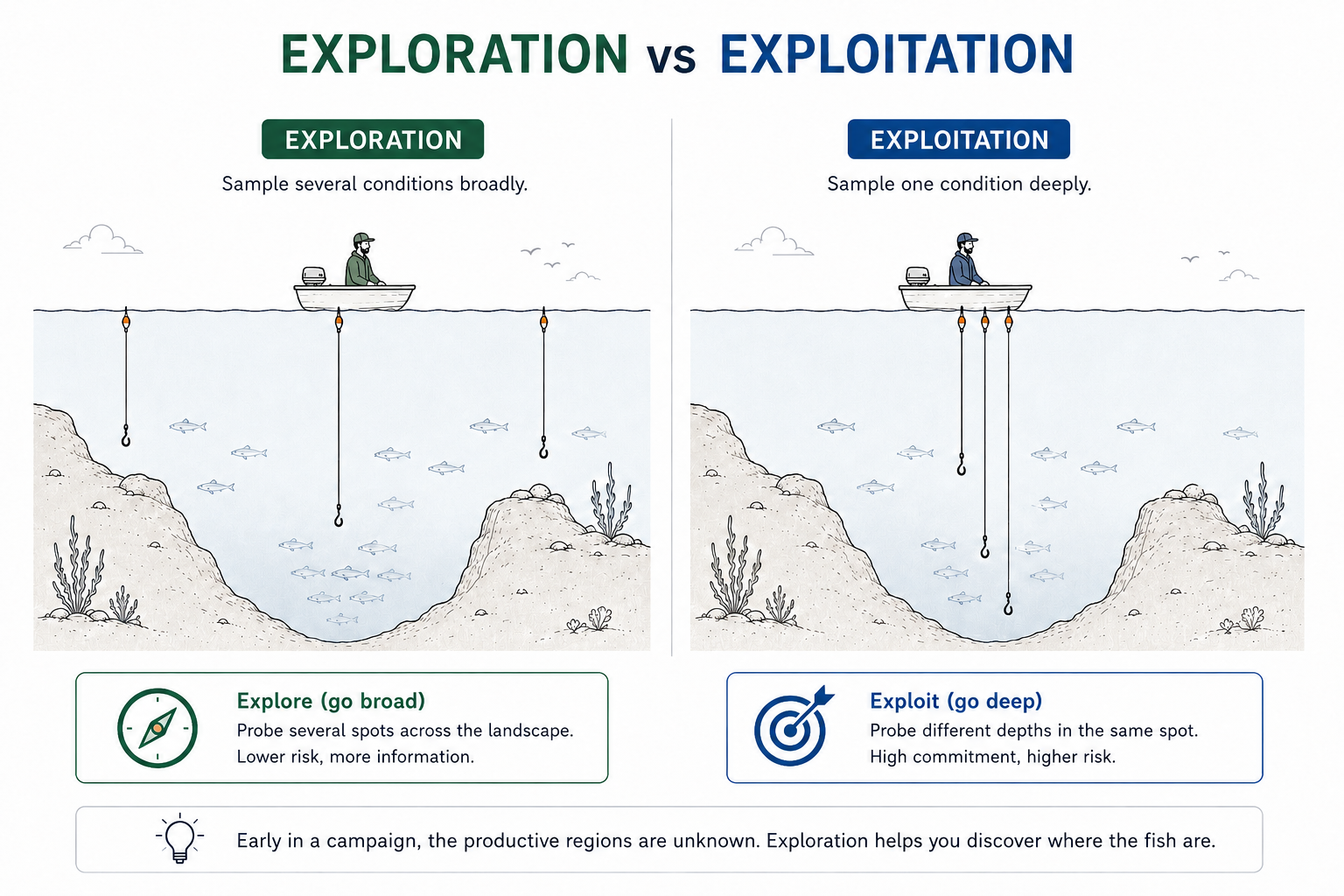
Exploitation versus exploration. Exploitation samples one region of design space intensively, while exploration distributes sampling across several distinct regions.
The same decision appears in generative design. A campaign might generate 1,000 candidates under one condition or 100 candidates under each of ten conditions. Early in a campaign, broad exploration is usually more informative because the productive regions are not yet known. Each condition must still be sampled deeply enough to distinguish a reproducible trend from stochastic variation.
3. Sequence design
Candidate geometry and amino-acid sequence are related but distinct design problems. When a model generates a backbone without assigning a complete sequence, an inverse-folding or sequence-design model is used to propose amino acids compatible with that structure.
In Volume 1, ProteinMPNN proposes sequences for the RFdiffusion backbones. Multiple sequences can be generated for each backbone, increasing sequence-level diversity without changing the intended geometry. The model primarily evaluates sequence–structure compatibility; it does not establish binding affinity, expression, or developability.
This stage creates an important model handoff. The backbone generator and sequence-design model solve different problems and do not necessarily share the same assumptions or failure modes. A geometry that appears plausible to the generative model may not support a sequence that robustly encodes that structure.
Tradeoff — sequence depth
Generating more sequences per structural hypothesis increases the chance of finding a sequence that recovers the intended fold, but it also increases downstream prediction and analysis costs. The appropriate depth depends on whether the campaign is still exploring structural space or refining a smaller number of promising geometries.
4. Structural recovery and plausibility
A designed sequence should be evaluated independently of the model that created it. Structure prediction can test whether the sequence appears to recover the intended fold, whether confidence is distributed consistently across the structure, and whether alternative conformations or disordered regions emerge.
In Volume 1, ESMFold predicts the mini-protein structure from sequence alone, without receiving the RFdiffusion backbone. The prediction is aligned to the intended design and evaluated using backbone RMSD, with per-residue confidence metrics such as pLDDT providing supporting evidence.
What structural recovery does and does not establish
Tradeoff — filter stringency
A strict structural-agreement threshold retains only designs with close model concordance but may eliminate borderline candidates that behave differently in the target-bound state. A looser threshold preserves diversity but admits greater uncertainty. Thresholds should reflect campaign purpose and downstream capacity rather than being treated as universal constants.
5. Interaction and interface assessment
For binders, enzymes, receptors, and other functional proteins, a plausible isolated fold is not enough. The campaign must evaluate whether the candidate engages the intended molecular partner in a useful geometry.
Relevant analyses may include residue–residue contacts, buried surface area, hydrogen bonds, salt bridges, hydrophobic packing, hotspot coverage, shape complementarity, catalytic geometry, substrate positioning, or overlap with a known ligand or receptor interface.
In Volume 1, interface fingerprinting characterizes how each designed mini-protein engages PD-L1 and whether it recovers the intended target surface. These measurements are informative structural proxies, but their relationship to affinity and function remains noisy. They are most useful when interpreted together rather than optimized individually.
Tradeoff — portfolio diversity
The shortlist should be a portfolio, not simply the top rows of a score-sorted table. Preserving diversity across poses, interface geometries, sequence families, hotspot usage, and mechanistic hypotheses reduces the risk that every selected candidate shares the same hidden failure mode.
6. Physical and ensemble-based stress testing
Static predictions represent individual structural hypotheses. More expensive physical or ensemble-based methods can test whether those hypotheses remain plausible when the system is allowed to relax, fluctuate, or sample nearby conformations.
Molecular dynamics is one such method. After system preparation, solvation, energy minimization, and equilibration, trajectories can be analyzed for structural drift, compaction, interface-contact persistence, hydrogen-bond occupancy, hotspot retention, and changes in the relative orientation of interaction partners.
MD is useful for exposing obvious structural or interfacial instability that is not apparent in a single static model. It does not directly measure binding affinity, and its conclusions depend on the force field, starting structure, simulation protocol, trajectory length, and sampling achieved. In Volume 1, MD functions as a comparative stress test rather than a definitive validation method.
Tradeoff — depth versus breadth
Longer or more numerous simulations provide greater opportunity to observe instability and alternative states, but they restrict the number of candidates that can be evaluated. Shorter simulations support broader triage but may miss slower failure modes. Computational depth should generally increase as the candidate pool narrows.
7. Independent or orthogonal model assessment
Confidence is strengthened when a molecular hypothesis survives assessment by a method that does not simply inherit the assumptions or coordinates of the original design model. The second method need not be completely independent to be useful, but the information supplied to it and the task it performs should differ meaningfully.
In Volume 1, Boltz-2 predicts the binder–target complex from sequence without receiving the RFdiffusion-designed backbone or starting pose. The resulting complex can be compared with the original design using global pose agreement, interface RMSD, hotspot recovery, contact overlap, and model-confidence metrics such as ipTM.
Agreement between the two modeling paths strengthens the proposed structural mechanism. Disagreement identifies candidates whose binding geometry is model-dependent, weakly encoded by sequence, or otherwise uncertain.
Tradeoff — agreement versus novelty
Requiring close agreement between models produces a conservative shortlist, but it may also remove unconventional candidates that one model represents poorly. Model agreement is evidence to weigh, not an automatic pass–fail rule.
8. Consensus scoring and candidate prioritization
The final computational stage integrates evidence collected across the campaign. Because no single metric captures all aspects of candidate quality, prioritization should use a transparent scorecard rather than allowing one confidence score, energy estimate, or structural metric to dominate by default.
The scorecard should retain the underlying measurements so that the reason for each decision remains visible. Weighting choices are campaign-specific. They should reflect the intended function, modality, known biology, downstream assay capacity, and the types of uncertainty the team is most willing to tolerate.
Final selection should also preserve diversity. A useful experimental shortlist may include high-confidence consensus candidates, distinct interface geometries, sequence-diverse representatives, and a small number of higher-risk hypotheses with unusually strong upside.
Tradeoff — ranking versus interpretability
A single composite score makes candidates easy to sort but can conceal contradictory evidence and arbitrary weighting decisions. A more interpretable scorecard preserves component metrics and uses the final ranking as a decision aid rather than a claim of absolute molecular quality.
When should a campaign stop or change direction?
Campaigns need explicit stopping and pivot criteria. Without them, the team can continue generating candidates or repeating analyses without learning whether the underlying strategy is productive. A stopping decision is not always a declaration of success or failure. It may indicate that sufficient evidence has been collected to advance, redirect, or terminate a particular design hypothesis.
✓ Advancement criteria met
One or more candidates meet the predefined computational or experimental criteria required to advance to the next stage.
⊘ Planned space explored
The major conditions or hypotheses have been tested without producing suitable candidates. The team can change modality, revise the specification, redefine the target surface, or stop.
⏱ Resource boundary reached
The available time, compute, material, or budget has been consumed. The best-supported candidates advance, while the remaining uncertainties and next steps are documented.
⚠ Systematic failure detected
Candidates repeatedly exhibit the same failure mode, such as convergence on the wrong surface, poor structural recovery, or a developability liability. The strategy should be redesigned rather than repeated unchanged.
The pipeline is also an experimental object
Each campaign round should improve more than the molecules. It should improve the design and screening infrastructure itself. Structured outcomes reveal where computational filters are informative, where they are redundant, which stages consume disproportionate resources, and which measurements actually predict experimental success.
Once experimental data become available, thresholds and scorecard weights can be calibrated against observed expression, stability, binding, specificity, and function. A filter that appeared convincing in theory may prove uninformative. A metric initially treated as secondary may emerge as a useful early predictor. These relationships can only be learned when model inputs, parameters, intermediate outputs, and experimental outcomes remain linked.
The long-term product of the campaign is therefore not merely a set of candidates. It is a reusable decision system: a structured record of what was attempted, what survived each gate, what failed experimentally, and how that evidence should change the next round.
A computational pipeline should become more selective because it
learns, not merely because it accumulates more filters.
Additional models and metrics add value only when they contribute
distinct information, expose a known uncertainty, or improve the
allocation of experimental resources.
Next: apply the framework in Volume 1
The three foundation pages established the campaign-level principles: Design-Build-Test-Learn, target and modality selection, screening-funnel architecture, model handoffs, tradeoffs, candidate portfolios, and stopping criteria.
Volume 1 now applies those principles to a complete de novo mini-protein binder campaign against PD-L1. The first detailed page begins with target preparation, hotspot selection, RFdiffusion backbone generation, ProteinMPNN sequence design, and sequence-only fold recovery.
Begin Volume 1 → De Novo Mini-Binder Design